Current Projects
Research in the Hayes group involves the synthesis of inorganic molecules for application in new chemical transformations and catalysis. Six main projects (two collaborative) address this goal.
Low Coordinate f-Element Complexes
Due to the large size and extreme Lewis acidity of the f-elements, preparing well-behaved metal complexes is exceedingly challenging. We have successfully utilized both known ligands and custom-designed frameworks to support an array of low (4 and 5) coordinate f-element compounds. These unusual species represent powerful models for studying fundamental bonding and reactivity patterns. For example, preliminary reaction chemistry has proven to be diverse and includes C–H and N–H bond activations relevant to important catalytic transformations (e.g. hydroamination).
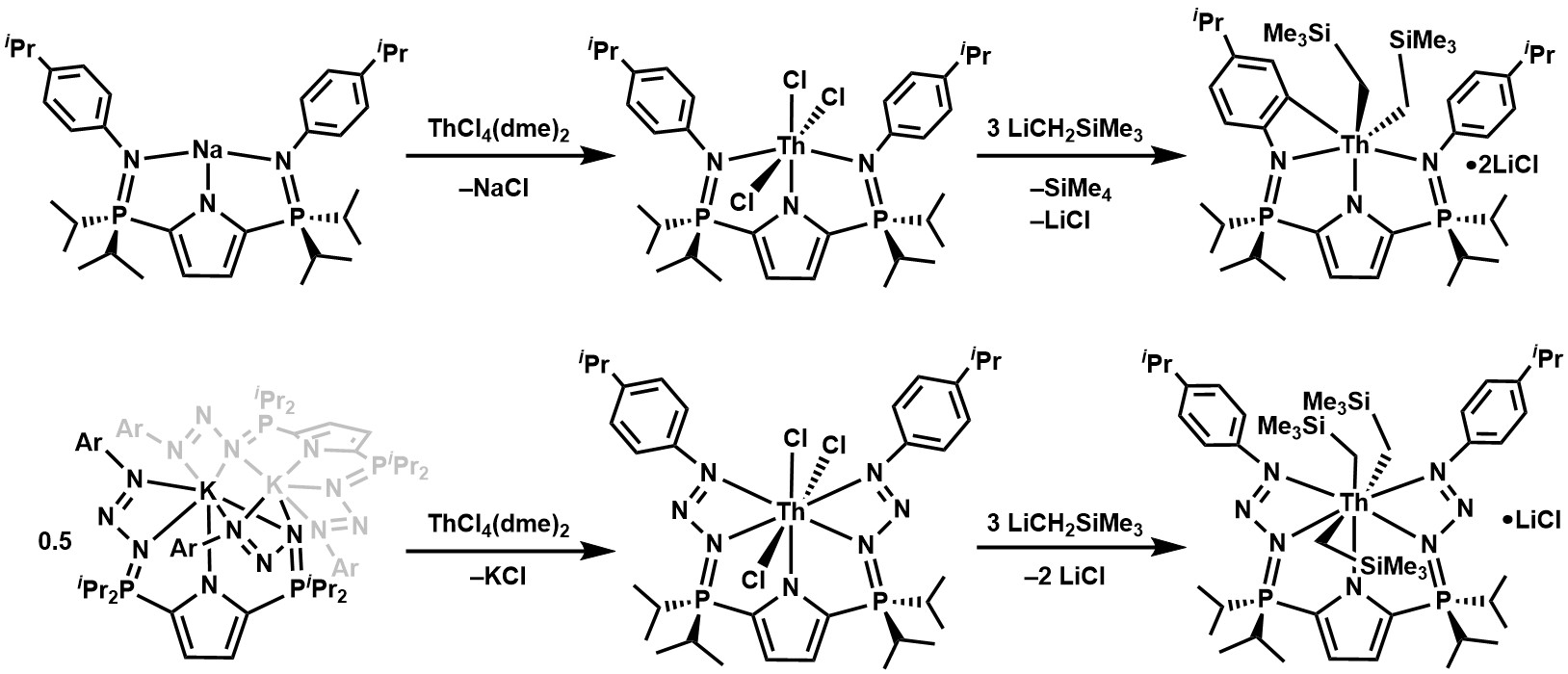
In a recent manuscript[1] we reported unprecedented coordination chemistry that expands our understanding of phosphazide (R3P–(N3)–R) and phosphinimine (R3P=N–R) functionalities. Notably, this work includes the first example of an actinide-stabilized phosphazide, as well as the first ligand to feature two phosphazide units that coordinate to a metal in a meridional fashion.
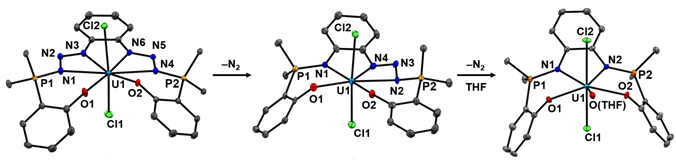
In our latest paper[2] on this topic we describe the synthesis and characterization of the thorium(IV) trialkyl complex LP=N3Th(CH2SiMe3)3 which is supported by a new pentadentate ligand that also features phosphazide moieties. In contrast to our uranium phosphazidosalen species (vide supra), these thorium diphosphazides are resistant to N2 loss at ambient temperature in solution. Furthermore, we demonstrate divergent reactivity between analogous phosphazide and phosphininmine-stabilized organothorium complexes; remarkably, the phosphinimine thorium trialkyl LP=NTh(CH2SiMe3)3 decomposes via cyclometallation of an N-aryl substituent, while LP=N3Th(CH2SiMe3)3 is thermally stable at ambient temperature in solution.
Small Molecule Functionalization Using Group 9 Pincer Complexes
A relatively new project in the aims to prepare low-coordinate group 9 complexes supported by NNN-pincer ligands. These species have been targeted because 14-electron, T-shaped intermediates, which are sterically and electronically unsaturated, are expected to participate in numerous bond activation and catalytic processes. Early experiments demonstrated that one of the phosphinimine donors in our Rh(I) complexes is hemilabile and can engage in cooperative metal-ligand activation of small molecules. For example, in the presence of excess H2, these complexes can catalytically hydrogenate a broad array of alkenes, including ethylene, under mild conditions.[1]
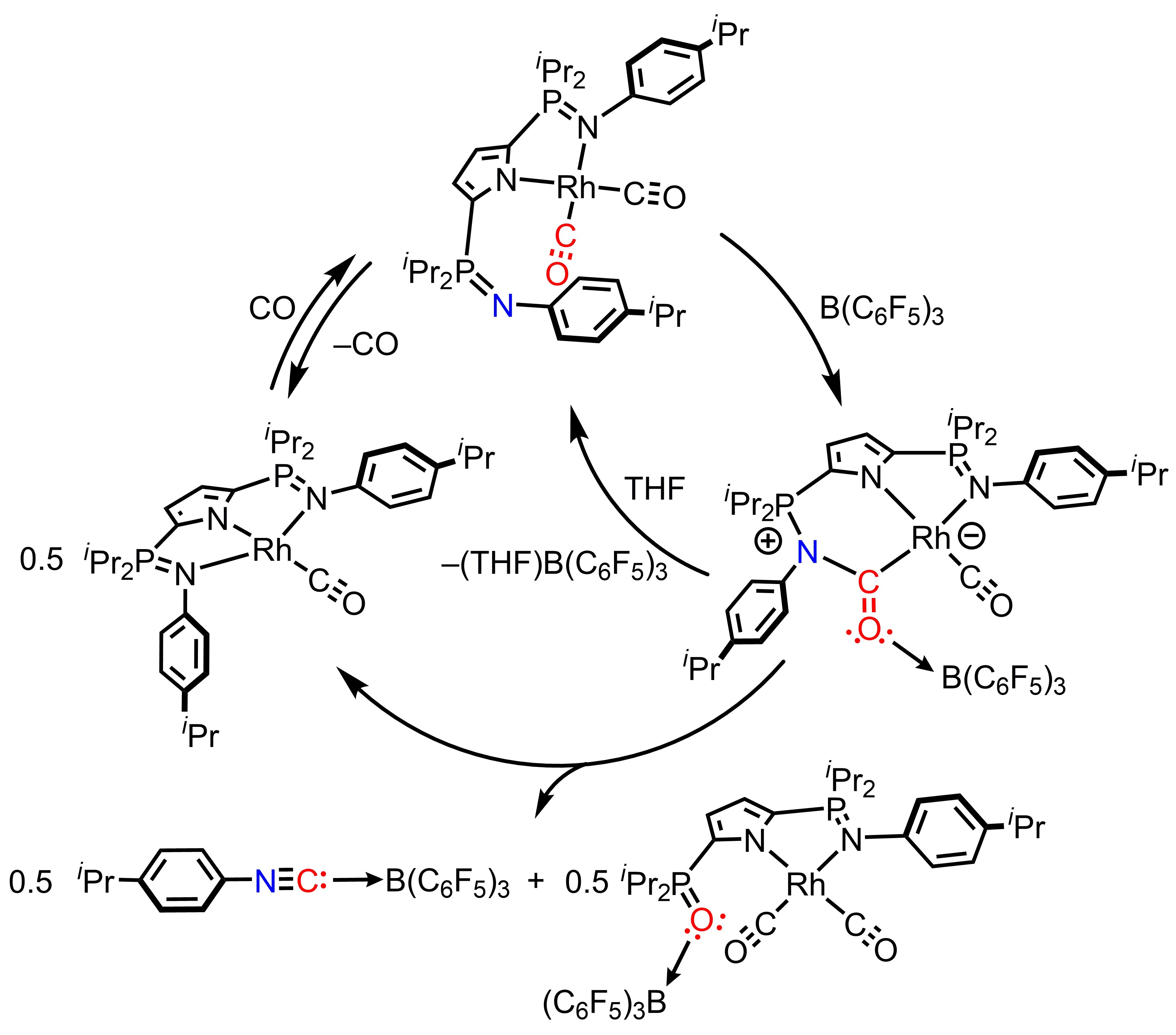
A slightly different ligand framework led to the discovery that reaction of LRh(CO)2 with the Lewis acidic borane B(C6F5)3, results in complete C–O bond scission.[2] This is notable because CO deoxygenation usually requires stoichiometric quantities of harsh reducing agents.
We have also prepared a family of neutral rhodium silylenes (LRh=SiR2) which are generated via the dehydrogenation of common hydrosilanes.[3] These complexes are important because they have been implicated as key intermediates in the rhodium catalyzed hydrosilation of ketones, but prior to our work no rhodium silylenes had been structurally characterized.
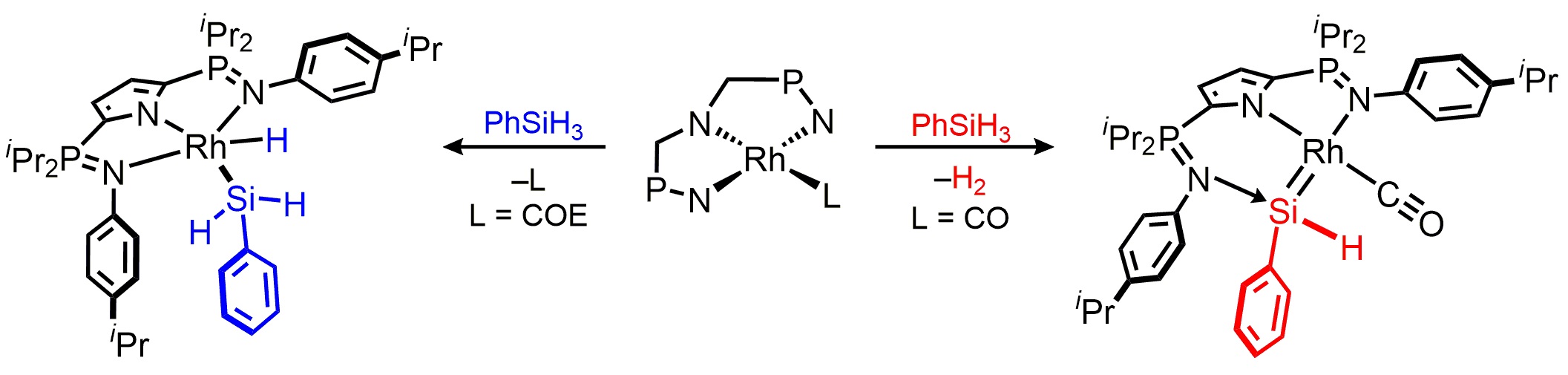
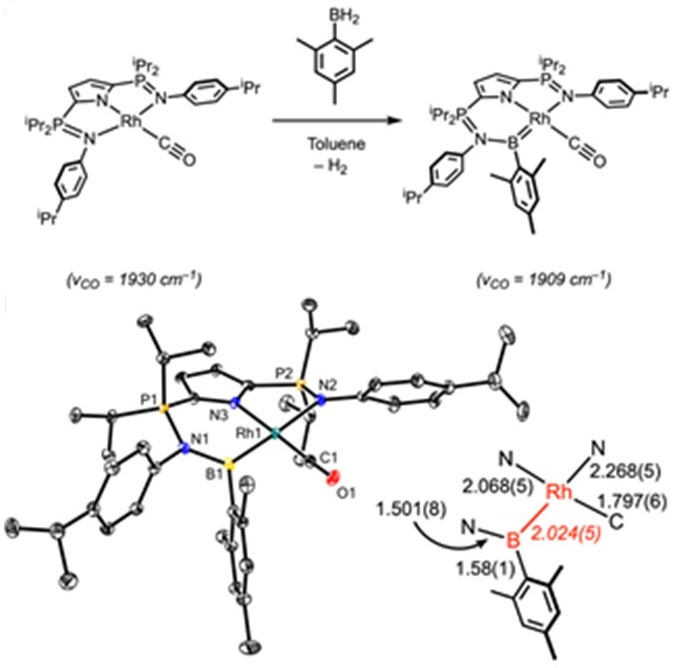
Most recently we discovered that a similar process permits the reversible dehydrogenation of primary boranes.[4]
Rare Earth Complexes Featuring Unusual Bonding Motifs
A separate thrust in the Hayes lab embodies the concept that species with unusual bonding or structure may lead to new chemical reactions and catalytic pathways. Specifically, this work aims to prepare rare earth complexes that feature metal main-group multiple-bonding (M=E; E = NR, PR, SiR2). Although such compounds are extremely rare they are often implicated in industrially relevant transformations including the metathesis of imines, aldehydes and carbodiimides and group transfer reactions. Complexes containing metal main-group mulitiple-bonds may also serve as models for intermediates in catalytic transformations, thus, providing a unique glimpse at otherwise fleeting structures.
Our efforts in this area include the design of new pincer platforms for supporting thermally sensitive metal complexes. For example, using a novel bis(phosphinimine)carbazole ligand, we prepared a mononuclear base-free dialkyl lutetium(III) complex.[1]
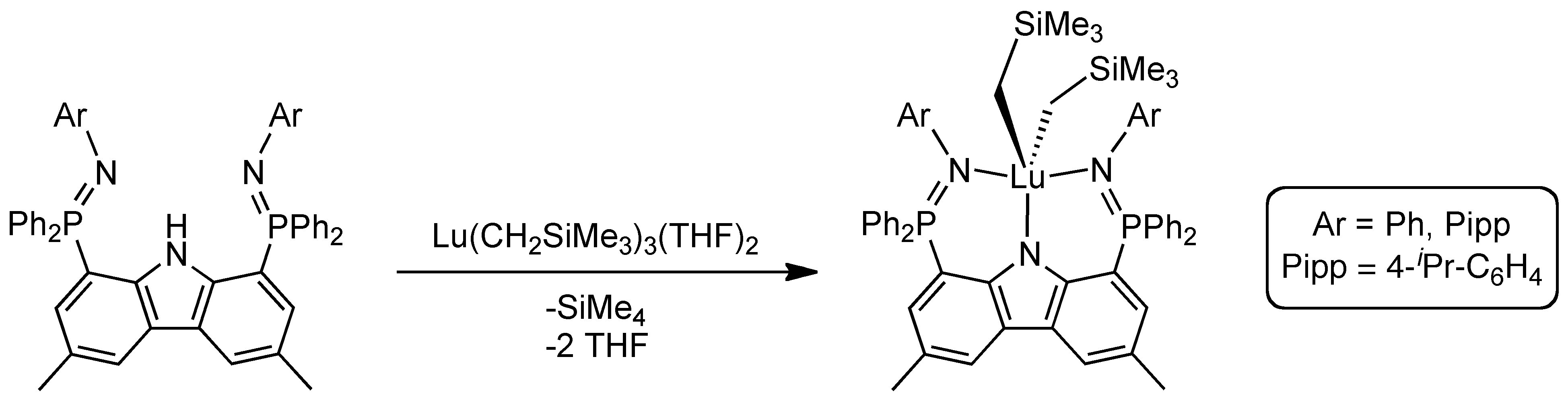
However, at temperatures above 0 °C, this compound undergoes two sequential intramolecular ortho-metallation processes.[1,2]
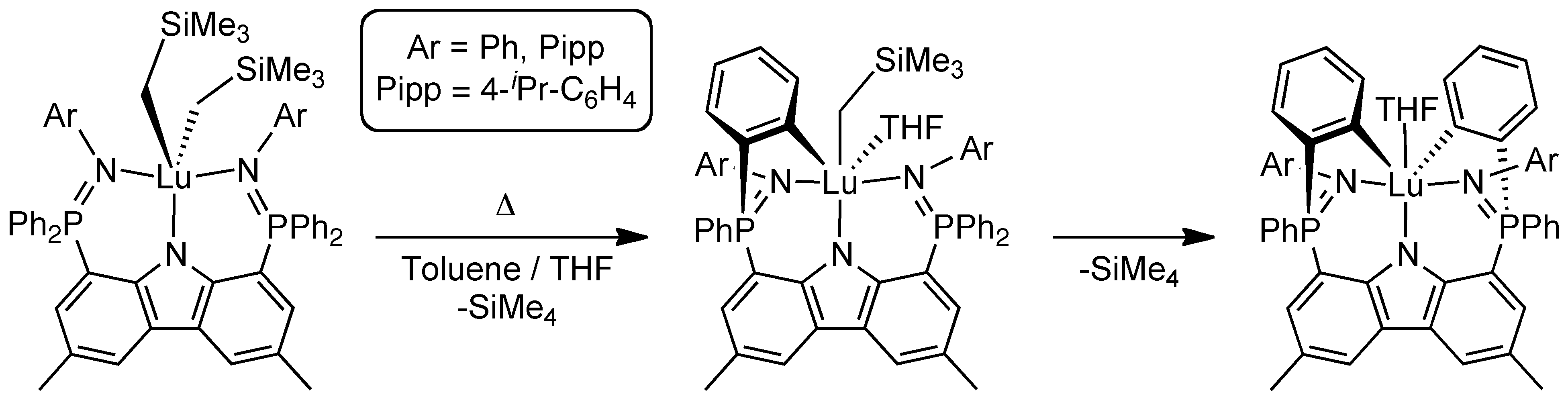
Further reactivity with substrates such as the bulky aniline Mes*NH2, affords a mixed anilide/aryl complex. This complex again is quite reactive and undergoes an intramolecular rearrangement to a different structural isomer. The nature of this rearrangement has been probed to determine if it proceeds through a transient lutetium imido (LLu=NR) intermediate. Kinetic and mechanistic (deuterium labelling) studies have demonstrated that the intramolecular rearrangement occurs via a unique direct metalation exchange process.[3]
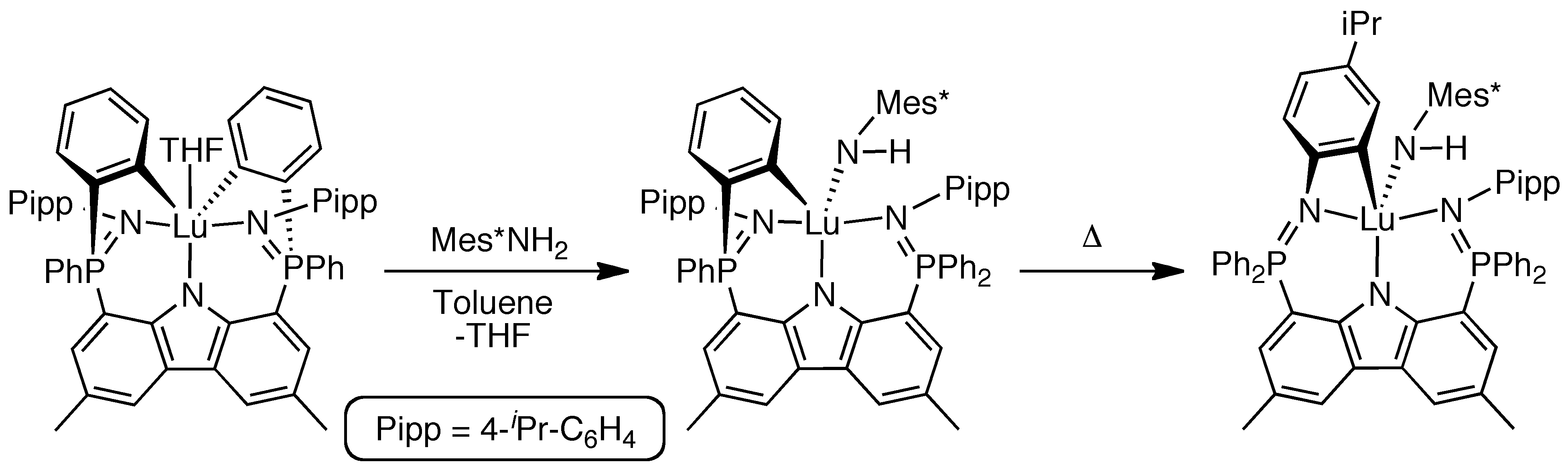
Newer ligand designs[4-6] ultimately generated complexes that are stable at 100 °C in solution for days.[7,8]
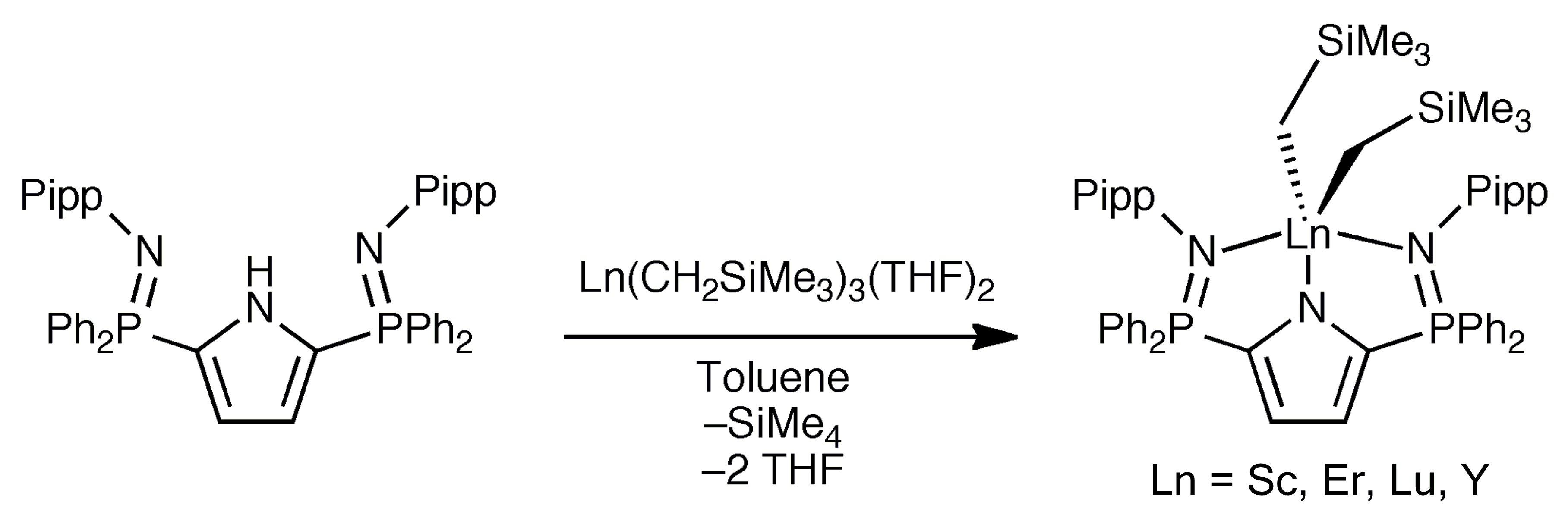
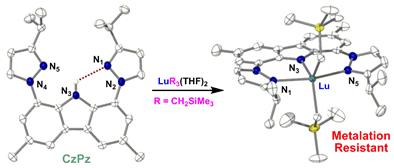
These and related complexes react readily with numerous small molecules, and hence, are promising catalyst candidates for a range of chemical reactions.
New Catalysts for the Synthesis of Green Polymers
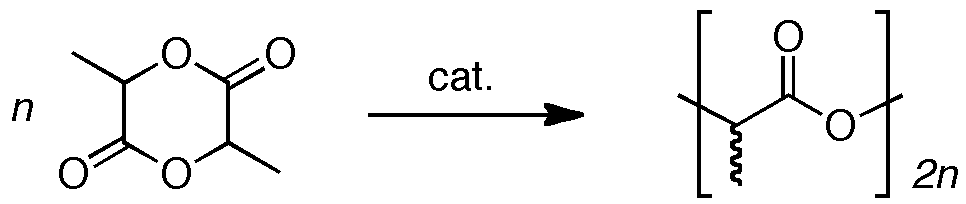
Our most developed research avenue tackles the challenge of preparing new materials that are both biodegradable and biocompatible. A particularly attractive alternative to conventional polyolefin materials is polylactide, PLA, which is generated by the ring-opening polymerization of lactide, the cyclic ester dimer of lactic acid. PLA is desirable because of its processability and useful macroscopic properties. The synthetic precursors to PLA are found in inexpensive renewable resources, such as beets and corn. Furthermore, the resultant polymers are biodegradable and have found applications in such areas as bulk packaging materials and the agricultural industry. It has also garnered interest from the medical community for its use in adsorbable sutures, as matrices for the slow release of pharmaceuticals, and polymer scaffolds for tissue engineering.
Despite these benefits, the industrial process for PLA synthesis involves a melt polymerization protocol in the presence of a metal containing catalyst. This method offers limited control over the molecular weights and stereochemistry of the final product; also, the metal catalyst remains embedded within the polymer. Our strategy focuses upon the development of discrete homogeneous lactide polymerization catalysts, specifically, group 2 and 12 metal alkyl, alkoxide and amido species.[1] Homogeneous systems are highly advantageous because they allow rational fine tuning of the steric and electronic environment of the metal centre.
Initial forays into this arena involved the preparation of zinc complexes of a neutral phosphinimine ligand featuring a dibenzofuran scaffold, which yielded complexes catalytically active in the polymerization of l-lactide at 100 °C.[2]
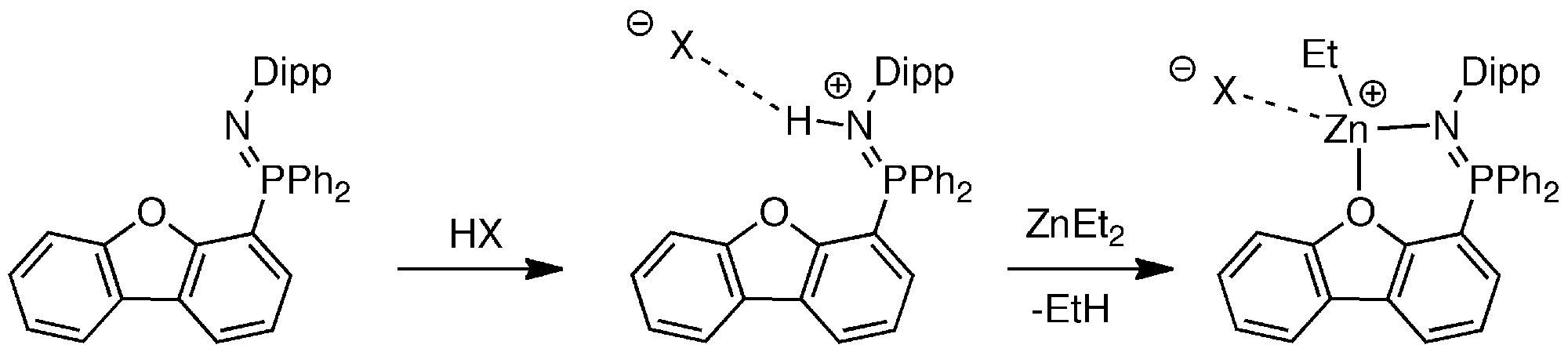
More recently, we have demonstrated that highly active cationic magnesium complexes of a neutral bis(phosphinimine) ligand were suitable for the preparation of poly(ε-caprolactone) at ambient temperature in several minutes.[3]
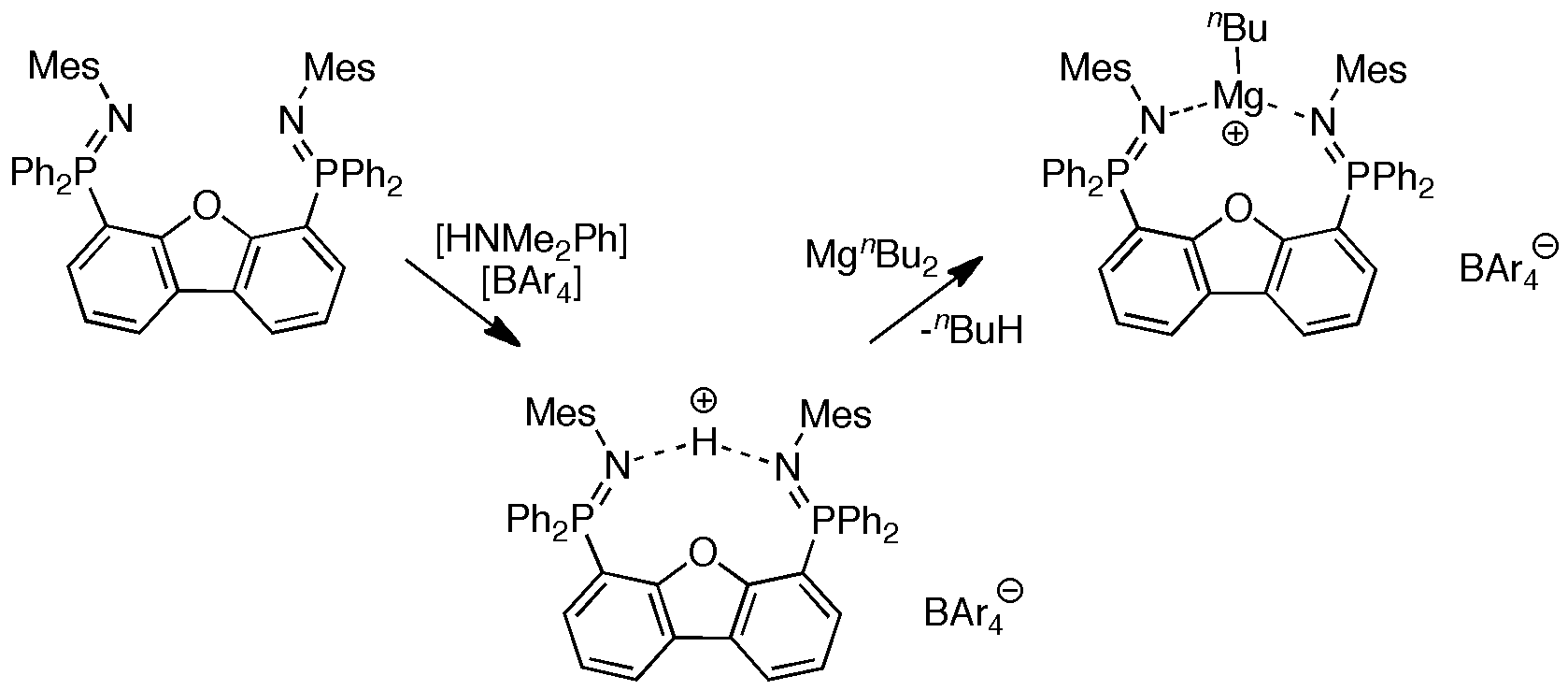
The preparation of analogous cationic organozinc complexes has also been achieved.[4,5] This knowledge was ultimately used to generate complexes with extremely high polymerization activity.[6,7]
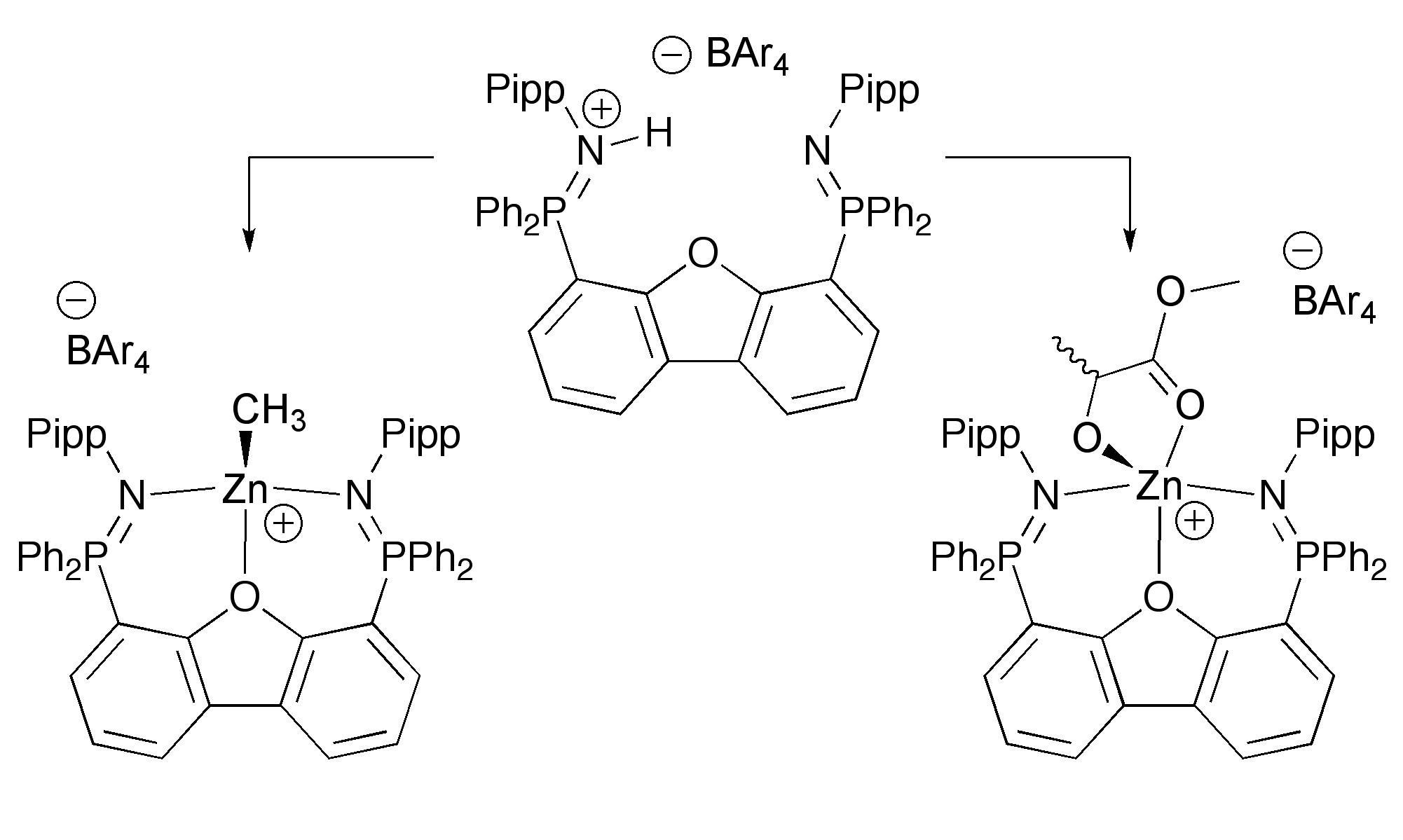
Other directions we have pursued include the preparation of chiral zinc catalysts supported by P-stereogenic ligands[8,9] and the installation of extreme ligand bulk in an attempt to overcome undesired tranesterification processes.[10] In summary, this project explores the largely uncharted organometallic chemistry of discrete cationic group 2 and 12 complexes[11] with the long term goal of contributing to the fundamental understanding of the production of environmentally friendly materials.
Collaborative Projects
Semisynthesis of Cannabinoids (with Prof. Igor Kovalchuk - Department of Biological Sciences)
Pharmaceutical and recreational cannabis strains typically contain large quantities of, Δ9-tetrahydrocannabinol (Δ9THC). Δ9THC exhibits numerous medicinal benefits, including pain reduction, sleep assistance, seizure treatment, etc. A disadvantage of Δ9THC, however, is that therapeutic doses often cause psychoactive effects. Also, high Δ9THC varieties must be grown in secure facilities. Conversely, high cannabidiol (CBD) varieties, which contain <0.3% Δ9THC, can be grown as industrial hemp in open fields.
Cannabinol (CBN) is closely related to Δ9THC, and shares many of its medicinal effects, especially anti-inflammatory properties. Furthermore, CBN is not psychoactive. Unfortunately, CBN is only found in low quantities in nature. Hence, we are seeking routes to large quantities of pure Δ9THC, CBN and other related cannabinoids without breaching government regulations. Over the past year these efforts have led to the development of high yielding semisynthetic methodologies for the synthesis, isolation, and purification of Δ9THC and CBN from CBD. Two patent applications have recently been issued to protect this technology, and together with our industrial partner, we are planning scale-up trials and targeting lesser known cannabinoids. Notably, preliminary studies indicate that several of our isolated cannabinoids are highly effective at selectively inhibiting the growth of breast cancer cells.
Polyphosphazenes (with Prof. Paul Hazenonk - Department of Chemistry)
A second polymer project studies inorganic polymers using solid-state NMR spectroscopy.[1] The work focuses specifically on a class of polymers known as poly(phosphazenes), which are hybrid inorganic-organic materials of great interest due to their potential applications in novel electronics, biomaterials and biodegradable plastics.
We have demonstrated that by using 31P magic-angle spinning (MAS) solid-state (SS) NMR spectroscopy, the complicated thermal ring-opening polymerization of hexachlorocyclotriphosphazene can be quantitatively probed to give information about reaction progress, chain propagation and cross-linking of the resultant polymer.[2]
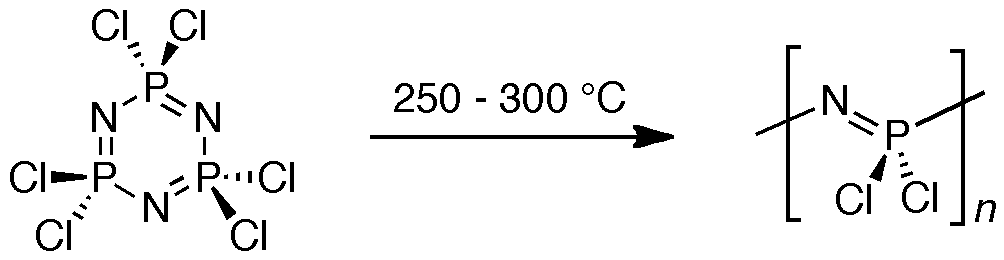
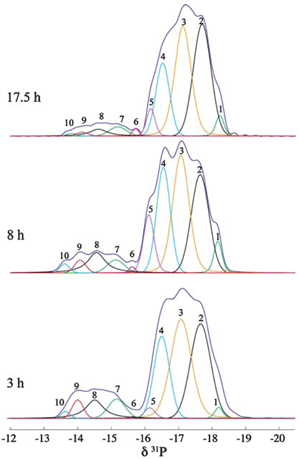
Deconvolution analysis of the SS 31P NMR spectra of a reaction mixture aliquot.
In a separate contribution, it was demonstrated that the detailed morphology of another polymer, poly[bis(trifluoroethoxy)phosphazene, could be probed using MAS SS NMR spectroscopy.[3] For instance, the degree of crystalline versus amorphous domains in a given sample can be determined with a specialized pulse sequence. Furthermore, it was demonstrated that SS NMR is a viable technique for assigning specific crystalline domains (e.g. α-, β- or γ-phases) and may prove to be a valuable complementary technique to X-ray diffraction methods.





