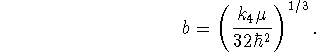The fundamental spectrum is centered on
![]() ; these lines are due to transitions during
which the vibrational quantum number is changed by
; these lines are due to transitions during
which the vibrational quantum number is changed by ![]() .
The first overtone is centered on
.
The first overtone is centered on
![]() (corresponding to transitions during which the
vibrational quantum number changes by 2)
and additional overtones (of decreasing
intensity and similar structure)
are potentially observable at
(corresponding to transitions during which the
vibrational quantum number changes by 2)
and additional overtones (of decreasing
intensity and similar structure)
are potentially observable at ![]() for
integer values of j.
The intensity of each overtone is lower than that of the
previous because overtones violate the harmonic oscillator selection
rule which is approximately obeyed for vibrational states.
Each line corresponds to a transition from a different rotational
state.
Lines to the left (high-energy) side of the centre correspond to
transitions during which the rotational quantum number increases.
Lines lower in energy than the centre of the fundamental or overtone
correspond to transitions during
which the rotational quantum number decreases.
for
integer values of j.
The intensity of each overtone is lower than that of the
previous because overtones violate the harmonic oscillator selection
rule which is approximately obeyed for vibrational states.
Each line corresponds to a transition from a different rotational
state.
Lines to the left (high-energy) side of the centre correspond to
transitions during which the rotational quantum number increases.
Lines lower in energy than the centre of the fundamental or overtone
correspond to transitions during
which the rotational quantum number decreases.
Let's start by adding up the orbital angular momenta. The quantum number for the total orbital angular momentum can be anything between 1-0 and 1+0. In other words, this quantum number can only be 1.
The total spin quantum number can be either ![]() or
or
![]() , i.e. either 0 or 1.
, i.e. either 0 or 1.
If the total spin quantum number is zero, the total angular momentum quantum number must be 1. If the total spin quantum number is 1, the total angular momentum quantum number can be any number between 1-1 and 1+1 in steps of 1. Thus the possible values of the total angular momentum quantum number are 0, 1 and 2.
On the timescale of motion of the electrons, nuclear motion is exceedingly slow. Thus nuclear-nuclear repulsion terms are effectively constant. Furthermore, since the nuclear wavefunction is relatively strongly localized, nuclear kinetic energy terms are zero almost everywhere. We can therefore reduce the electronic Hamiltonian to
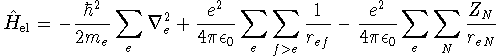
where lower case subscripts label electrons and upper case subscripts label nuclei. The dependence of the electronic wavefunctions and energies on the nuclear positions enters parametrically through the electron-nuclear attraction terms. We solve the electronic problem for several different nuclear geometries (different bond lengths and angles). For each geometry, we obtain a series of electronic energies. Each electronic state therefore corresponds to an energy hypersurface parameterized by the nuclear coordinates.
After solving the electronic problem, the molecular Hamiltonian becomes
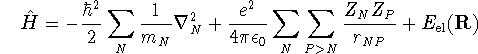
where ![]() is the vector of nuclear coordinates. If we
add the last two terms, we get the effective potential energy
(
is the vector of nuclear coordinates. If we
add the last two terms, we get the effective potential energy
( ![]() )
experienced by the nuclei. Thus nuclear motion obeys the
Schrödinger equation
)
experienced by the nuclei. Thus nuclear motion obeys the
Schrödinger equation
![]()
where
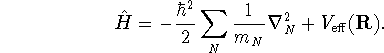
> assume(b>0); > avgK := -hbar^2/(2*mu)*sqrt(b/Pi) > *int(exp(-b*x^2/2)*diff(exp(-b*x^2/2),x$2), > x=-infinity..infinity);
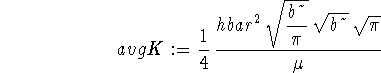
> simplify(avgK);
![]()
I carried out the next step using an advanced Maple command
(subs()), but this could equally well have been done by hand.
> subs(b=sqrt(k*mu)/hbar,");
![]()
This is ![]() , or half the ground state energy.
Incidentally, this is an example of the virial theorem. For
potentials which are homogeneous functions of their arguments (functions
for which
, or half the ground state energy.
Incidentally, this is an example of the virial theorem. For
potentials which are homogeneous functions of their arguments (functions
for which ![]() ), the
average kinetic energy is always a simple multiple of the total energy.
), the
average kinetic energy is always a simple multiple of the total energy.
Let's start by defining the wavefunction:
> phi:=x->exp(-b*x^2);
![]()
The variational energy is
![]() .
Let's compute it one part at a time:
.
Let's compute it one part at a time:
> top:=int(phi(x)*(-hbar^2/(2*mu)*diff(phi(x),x$2) > +k4*x^4/24*phi(x)),x=-infinity..infinity);
![]()
> bottom:=int(phi(x)^2,x=-infinity..infinity);
![]()
> Evar := simplify(top/bottom);
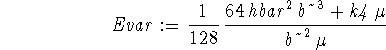
To find the best value of b, we minimize the variational energy with
respect to b:
> sol:=solve(diff(Evar,b)=0,b);
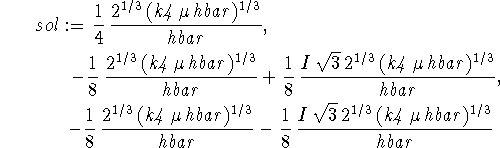
Only the first of these solutions is physically reasonable.
Again, I'm going to use subs() to do the work, although you
probably worked out these parts of the problem by hand.
subs(b=sol[1],Evar);
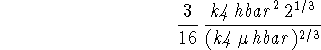
The normalization factor is ![]() .
.
> sqrt(1/bottom);

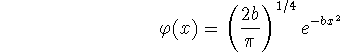
with