> psi2s := (r,theta,phi) -> 1/8*sqrt(2/(Pi*a0^3))*(2-r/a0)*exp(-r/(2*a0));
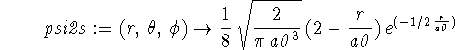
> psi3s := (r,theta,phi) -> 1/243*sqrt(3/(Pi*a0^3))*(27-18*r/a0+2*(r/a0)^2) *exp(-r/(3*a0));
Maple will need to know the sign of a0 to solve the problems in
this assignment.
> assume(a0>0);
In the 2s state,
> int(int(int( evalc(conjugate(psi2s(r,theta,phi))*r*psi2s(r,theta,phi)) *r^2*sin(theta),theta=0..Pi),phi=0..2*Pi),r=0..infinity);
![]()
evalc() and conjugate() are not
strictly required in this and the following
case since the wavefunctions are real.
In the 3s state,
> int(int(int( evalc(conjugate(psi3s(r,theta,phi))*r*psi3s(r,theta,phi)) *r^2*sin(theta),theta=0..Pi),phi=0..2*Pi),r=0..infinity);
![]()
As discussed in introductory chemistry courses, the shells get bigger
(the electrons spend more time away from the nucleus) as the principal
quantum number increases. The increase in average distance from the
nucleus ( ![]() ) reflects this.
) reflects this.
> rpd3s := r -> N3s*r^2*(27-18*r/a0+2*(r/a0)^2)^2 *exp(-2*r/(3*a0));
![]()
N3s is a normalization factor which we must determine:
> solve(int(rpd3s(r),r=0..infinity)=1,N3s);
![]()
> N3s:=";
![]()
Now, for the ![]() state (
state ( ![]() has an identical r.p.d.):
has an identical r.p.d.):
> rpd3d2 := r -> N3d2*r^2*(r/a0)^4*exp(-2*r/(3*a0));
![]()
> solve(int(rpd3d2(r),r=0..infinity)=1,N3d2);
![]()
> N3d2:=";
![]()
> plot(subs(a0=1,rpd3s(r)),r=0..40);
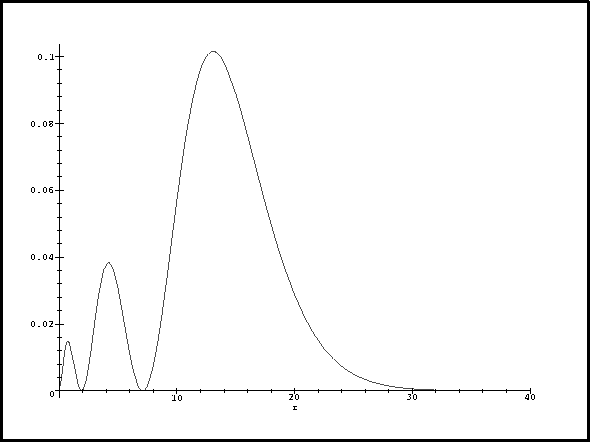
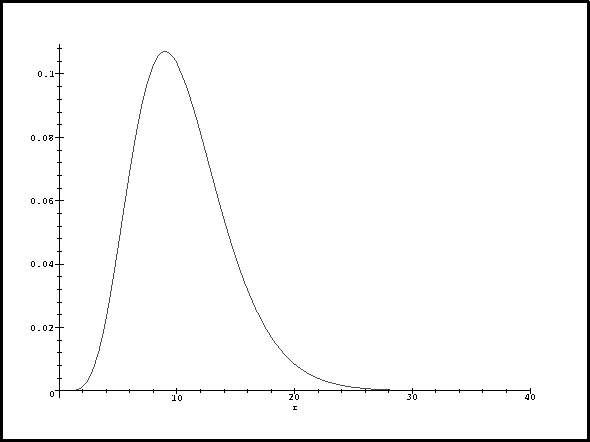
For the ![]() state:
state:
> plot(subs(a0=1,rpd3d2(r)),r=0..40);
> solve(rpd3s(r)=0,r);
![]()
![]()
> rn1:="[3];
![]()
> rn2 := ""[4];
![]()
> rn0:=0;
![]()
> int(rpd3s(r),r=rn1..infinity);
![]()
> evalf(");
![]()